
Our Research Lines
AI-Enabled Computational Drug Discovery
The large amount of available chemical and biological data allows us to leverage diverse predictive models for chemical and biological properties. We thus aim to explore static protein conformations (through AlphaFold2/3), dynamical protein states (through classical and quantum machine learning force fields) or small molecule structurs and properties (through generative models).
Computational Molecular Biophysics
Molecular simulations serve as a computational microscope for revealing dynamic processes that are unable to being captured by other methods. Understanding the role of protein dynamics in drug-ligand interactions and corresponding conformational changes can help us design better molecules with therapeutic applications. We leverage on enhanced sampling techniques such as Hamiltonian Replica-Exchange, AI-guided Metadynamics, and Markov State Models to explore the conformational changes of proteins and how these are linked to health and disease.
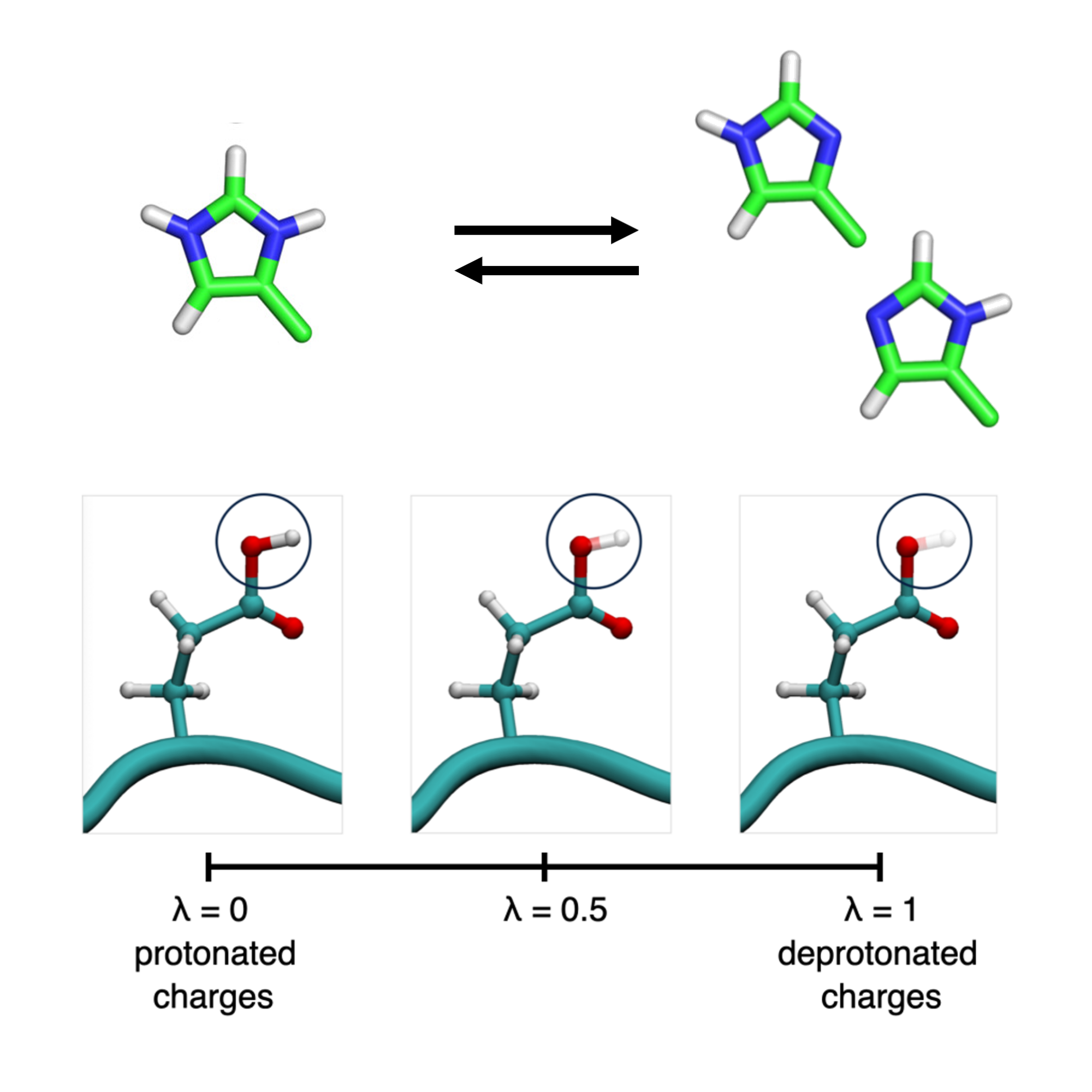
Protonation events in Chemistry
Together with Prof. Czodrowski, we are in understanding the role of proton transfer in small molecules, such as tautomeric processes and pKa changes.
Our Publications
- Small-molecule modulators of TRMT2A decrease PolyQ aggregation and PolyQ-induced cell death
Read more
- Design, synthesis, and in silico multitarget pharmacological simulations of acid bioisosteres with a validated in vivo antihyperglycemic effect
Read more
- D-Peptide Builder: A Web Service to Enumerate, Analyze, and Visualize the Chemical Space of Combinatorial Peptide Libraries
Read more
- Molecular Dynamics-Assisted Interpretation of Experimentally Determined Intrinsically Disordered Protein Conformational Components
Read more
- Antiallodynic effect of PhAR-DBH-Me involves cannabinoid and TRPV1 receptors
Read more
Small-molecule modulators of TRMT2A decrease PolyQ aggregation and PolyQ-induced cell death
Abstract
Polyglutamine (polyQ) diseases are characterized by an expansion of cytosine-adenine-guanine (CAG) trinucleotide repeats encoding for an uninterrupted prolonged polyQ tract. We previously identified TRMT2A as a strong modifier of polyQ-induced toxicity in an unbiased large-scale screen in Drosophila melanogaster. This work aimed at identifying and validating pharmacological TRMT2A inhibitors as treatment opportunities for polyQ diseases in humans. Computer-aided drug discovery was implemented to identify human TRMT2A inhibitors. Additionally, the crystal structure of one protein domain, the RNA recognition motif (RRM), was determined, and Biacore experiments with the RRM were performed. The identified molecules were validated for their potency to reduce polyQ aggregation and polyQ-induced cell death in human HEK293T cells and patient derived fibroblasts. Our work provides a first step towards pharmacological inhibition of this enzyme and indicates TRMT2A as a viable drug target for polyQ diseases.
Methods
The crystal structure of the human TRMT2A RRM (aa 69 – 147) was determined using a cloning, expression, and purification process. The human TRMT2A RRM was expressed in Rosetta E. coli cells in LB medium, and the lysate was clarified by centrifugation, loaded onto a His-FFTrap column, and washed with 10 column volumes (CV) of His-A buffer, followed by a 10 CV wash with His-B buffer. The purified protein was concentrated to 8 mg/ml by ultrafiltration. Protein concentrations were determined by measurement of the A280. Aliquots were flash-frozen in liquid nitrogen and stored at −80 °C.
Crystallization experiments for the RRM domain were performed at the X-ray Crystallography Platform at Helmholtz Zentrum, München. Crystallization screening was done at 292 K using 8.0 mg/ml of protein with a nanodrop dispenser in sitting-drop 96-well plates and commercial screens. Crystals appeared after one week and were big enough for X-ray diffraction experiments. The best data set used for refinement was collected for a crystal grown in 0.2 M cesium sulfate and 2.2 M ammonium sulfate.
The structure of the RRM with a resolution of 2.02 Å was solved with the Auto-Rickshaw pipeline using the sequence as input. For the molecular replacement step followed by several cycles of automated model building and refinement, the Auto-Rickshaw pipeline invoked the following X-ray crystallography software: MORDA, CCP4, SHELXE, BUCCANEER, RESOLVE, REFMAC5, and PHENIX. Model rebuilding was performed in COOT. Initial refinement was done with the 2.02 Å dataset in REFMAC5 [32] using the maximum–likelihood target function. Further refinement was done with another dataset with a higher resolution of 1.23 Å. The stereochemical analysis of the final model was done in PROCHECK [35] and MolProbity [35]. The final model is characterized by R/Rfree factors of 12.60 / 18.90%. Atomic coordinates and structure factors for the 2.0 Å and 1.23 Å structures have been deposited to the Protein Data Bank under accession codes 7NTN and 7NTO, respectively.
In-silico analyses of the crystal structure included binding site detection using agglomerative Euclidean distance clustering on the crystallographic water (oxygen atom) clusters, alignments performed with ClustalW [36], conservation analyses performed with AL2CO [37], and binding site detection using DoGSiteScorer [38], the transient site discovery, the machine learning-based CryptoSite approach, and TRAPP [40]. TRAPP is a framework to generate and analyze large-scale secondary structure rearrangements on short molecular dynamics timescales, e.g., by disrupting sidechain atoms with short pulses (L-RIP). The system temperature is controlled by a Langevin thermostat to ensure a constant average temperature. In parallel, the researchers investigated ligand-mediated conformational changes and putative allosteric effects in TRMT2A RRM with AlloPred [42].
Molecular dynamics simulations were performed using Maestro (Schrödinger Release 2016–2) and tleap (AmberTools 17) to prepare the protein structure for explicit solvent molecular dynamics simulations. The domain was embedded in a truncated octahedral TIP3P water box with a buffer of 15 Å, resulting in about 6690 water molecules. Neutralizing the system was achieved by introducing nine randomly placed Cl- counterions, placed with the addion2 application, to minimize perturbations of the system by adding charges too close to the protein. After minimization with harmonic restraints on protein-heavy atoms, the system was gradually heated from 100 to 300 K over 200 ps in NVT ensemble. A density equilibration over 1 ns was performed, followed by free simulations of the systems in NpT ensemble over 50 ns to ensure reasonable equilibration of the simulated systems. Production runs were carried out at 300 K using the Langevin thermostat [46] at 1.0 bar with an 8.0 Å nonbonded cutoff.
Design, synthesis, and in silico multitarget pharmacological simulations of acid bioisosteres with a validated in vivo antihyperglycemic effect
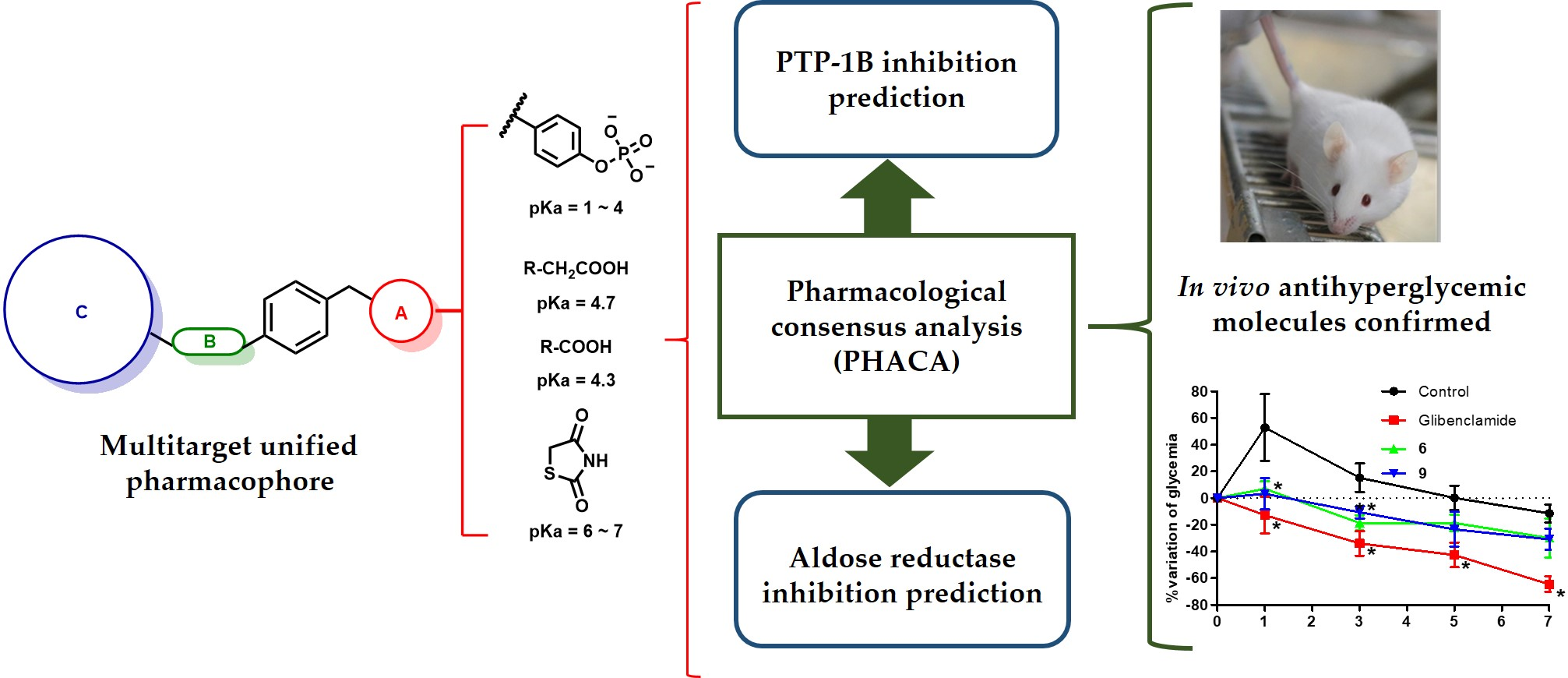
Abstract
Substituted phenylacetic (1–3), phenylpropanoic (4–6), and benzylidenethiazolidine-2,4-dione (7–9) derivatives were designed according to a multitarget unified pharmacophore pattern that has shown robust antidiabetic activity. This bioactivity is due to the simultaneous polypharmacological stimulation of receptors PPARα, PPARγ, and GPR40 and the enzyme inhibition of aldose reductase (AR) and protein tyrosine phosphatase 1B (PTP-1B). The nine compounds share the same four pharmacophore elements: an acid moiety, an aromatic ring, a bulky hydrophobic group, and a flexible linker between the latter two elements. Addition and substitution reactions were performed to obtain molecules at moderated yields. In silico pharmacological consensus analysis (PHACA) was conducted to determine their possible modes of action, protein affinities, toxicological activities, and drug-like properties. The results were combined with in vivo assays to evaluate the ability of these compounds to decrease glucose levels in diabetic mice at a 100 mg/kg single dose. Compounds 6 (a phenylpropanoic acid derivative) and 9 (a benzylidenethiazolidine-2,4-dione derivative) ameliorated the hyperglycemic peak in a statically significant manner in a mouse model of type 2 diabetes. Finally, molecular dynamics simulations were executed on the top performing compounds to shed light on their mechanism of action. The simulations showed the flexible nature of the binding pocket of AR, and showed that both compounds remained bound during the simulation time, although not sharing the same binding mode. In conclusion, we designed nine acid bioisosteres with robust in vivo antihyperglycemic activity that were predicted to have favorable pharmacokinetic and toxicological profiles. Together, these findings provide evidence that supports the molecular design we employed, where the unified pharmacophores possess a strong antidiabetic action due to their multitarget activation.
Methods
The study used commercially available reagents and performed experiments on ICR male mice. The animals were housed in animal cages with 12 hours of light and 12 hours of dark, and maintained in a 25°C environment. All animal experiments were conducted according to protocols approved by the Mexican government.
The induction of diabetes was performed using Nicotinamide (NA) and streptozotocin (STZ) as drugs. Mice with blood glucose levels over 150 mg/dL were used for the in vivo assay. The acute antidiabetic assay was performed using a previous methodology. Blood glucose levels were measured at 0, 1, 3, 5, and 7 hours using a commercial glucometer.
Crystal structures of proteins PTP-1B and AR were downloaded from PDB and manually curated using Maestro 12.1 with the Protein Preparation Wizard from the Schrodinger suite 2019-4. The hydrogen-bond network and rotameric conformations were optimized and restrained minimization was performed.
Molecular dynamics simulations were performed using Desmond with the OPLS3e forcefield. The complexes were prepared with the System Builder Utility in a 10 Å buffered rhombic dodecahedron box, and the complexes were minimized using the steep-descent algorithm followed by the Broyden–Fletcher–Gold–Farb–Shanno (LBFGS) method in three stages. Equilibration was carried out in several steps, including Brownian dynamics for 250 ps, simulation on the NVT ensemble, and equilibration on the NPT ensemble.
After the system was ready for production, 300 ns of simulation were performed under an NPT ensemble at 1 bar and 300 K using the Martyna–Tuckerman–Klein (MTK) barostat and the Nosé–Hoover thermostat. Electrostatic forces were calculated with the smooth PME method using a 9 Å cut-off, while constraints were enforced with the M-SHAKE algorithm. Integration was done every 2 fs, with a recording interval of 500 ps. Quality analysis of the simulation and other trajectory analyses were carried out using tools implemented in the Maestro 12.1 and VMD.
The study focuses on ensemble docking of two drug-bound receptors in the holo state, using Glide 8.4 with the XP methodology and the OPLS3e forcefield. The first six clusters, covering approximately 60% of the sampled conformational space, were recovered. Docking procedures were performed on the six protein conformations with nine compounds and the corresponding cocrystallized inhibitor. A grid was created around the binding pocket to allow the ligand to explore diverse binding poses.
Results for each compound were pooled and clustered following the ligand heavy atoms. A simple data fusion approach was followed, selecting the pose within the most populated cluster and assigning the docking score based on the lowest value within the most populated cluster. Validation of the docking protocol was carried out by redocking the respective cocrystallized ligands.
An a priori pharmacological consensus analysis (PHACA) was performed using results from several computational tools. Pharmacodynamic properties were calculated from the output score obtained with Glide XP, while pharmacokinetic properties were acquired using AdmetSAR and SwissADME software. Toxicological profiles were obtained from AdmetSAR and ACD/Tox Suite version 2.95.
The pharmacological consensus analysis was visualized using a color code indicating chances of the compound having drug-like properties: green (very satisfactory), yellow (satisfactory), and red (unsatisfactory). Three properties were considered for each point, and the compounds were classified based on their satisfaction with these properties.
D-Peptide Builder: A Web Service to Enumerate, Analyze, and Visualize the Chemical Space of Combinatorial Peptide Libraries
Abstract
Peptide-based drug discovery is re-gaining attention in drug discovery. Similarly, combinatorial chemistry continues to be a useful technique for the rapid exploration of chemical space. A current challenge, however, is the enumeration of combinatorial peptide libraries using freely accessible tools. To facilitate the swift enumeration of combinatorial peptide libraries, we introduce herein D-Peptide Builder. In the current version, the user can build up to pentapeptides, linear or cyclic, using the natural pool of 20 amino acids. The user can use non- and/or N-methylated amino acids. The server also enables the rapid visualization of the chemical space of the newly enumerated peptides in comparison with other libraries relevant to drug discovery and preloaded in the server. D-Peptide Builder is freely accessible at http://dpeptidebuilder. quimica.unam.mx:4000/. It is also accessible through the open D-Tools platform (DIFACQUIM Tools for Chemoinformatics https://www.difacquim.com/d-tools/).
Molecular Dynamics-Assisted Interpretation of Experimentally Determined Intrinsically Disordered Protein Conformational Components
Abstract
Mass spectrometry and single molecule forcemicroscopy are two experimental approaches able to providestructural information on intrinsically disordered proteins (IDPs).These techniques allow the dissection of conformationalensembles in their main components, although at a low-resolutionlevel. In this work, we interpret the results emerging from theseexperimental approaches on human alpha synuclein (AS) byanalyzing a previously published 73μs-long molecular-dynamics(MD) simulation of the protein in explicit solvent. We furthercompare MD-based predictions of single molecule Försterresonance energy transfer (smFRET) data of AS in solution withexperimental data. The combined theoretical and experimentaldata provide a description of AS main conformational ensemble, shedding light into its intramolecular interactions and overallstructural compactness. This analysis could be easily transferred to other IDPs.
Methods
The study analyzed MD data from DESRES, a simulated molecule with residues 1-14. The data was sampled every 10 ns, recovering 7,320 structures. Population uncertainty was estimated through a bootstrap analysis with 50 iterations. Hydrogen-bond and salt bridge contacts were computed using the Hydrogen Bond and Salt Bridges modules of the VMD code, while radii of gyration were computed using the gmx gyrate code. The Solvent-Accessible Surface Area (SASA) was computed using the Shrake-Rupley algorithm in MDTraj. The Accessible Solvent Area percentage was calculated from the mean values of each residue relative to the Maximum Accessible Solvent Area values reported. The collisional cross-section values were computed using the CoSIMS code. The smFRET efficiency (FE) was averaged along the DESRES trajectory. The gmx cluster code was used for structural clustering, while the AgglomerativeClustering module of the scikit-learn library in Python was used for feature clustering. The quality of the clustering parameters was established using the elbow method, Silhouette, and Calinski-Harabasz scores. Two sets of CCs were obtained, and the overlap between them was calculated using a dimensionality reduction projection. The experimental SASA was estimated from linear models based on the average charge state of conformers in native MS data.
Antiallodynic effect of PhAR-DBH-Me involves cannabinoid and TRPV1 receptors

Abstract
The antiallodynic effect of PhAR-DBH-Me was evaluated on two models of neuropathic pain, and the potential roles of CB1, CB2, and TRPV1 receptors as molecular targets of PhAR-DBH-Me were studied. Female Wistar rats were submitted to L5/L6 spinal nerve ligation (SNL) or repeated doses of cisplatin (0.1 mg/kg, i.p.) to induce experimental neuropathy. Then, tactile allodynia was determined, and animals were treated with logarithmic doses of PhAR-DBH-Me (3.2-100 mg/kg, i.p.). To evaluate the mechanism of action of PhAR-DBH-Me, in silico studies using crystallized structures of CB1, CB2, and TRPV1 receptors were performed. To corroborate the computational insights, animals were intraperitoneally administrated with antagonists for CB1 (AM-251, 3 mg/kg), CB2 (AM-630, 1 mg/kg), and TRPV1 receptors (capsazepine, 3 mg/kg), 15 min before to PhAR-DBH-Me (100 mg/kg) administration. Vagal stimulation evoked on striated muscle contraction in esophagus, was used to elicited pharmacological response of PhAR-DBH-ME on nervous tissue. Systemic administration of PhAR-DBH-Me reduced the SNL- and cisplatin-induced allodynia. Docking studies suggested that PhAR-DBH-Me acts as an agonist for CB1, CB2, and TRPV1 receptors, with similar affinity to the endogenous ligand anandamide. Moreover antiallodynic effect of PhAR-DBH-Me was partially prevented by administration of AM-251 and AM-630, and completely prevented by capsazepine. Finally, PhAR-DBH-Me decreased the vagally evoked electrical response in esophagus rat. Taken together, results indicate that PhAR-DBH-Me induces an antiallodynic effect through partial activation of CB1 and CB2 receptors, as well as desensitization of TRPV1 receptors. Data also shed light on the novel vanilloid nature of the synthetic compound PhAR-DBH-Me.
2 METHOLOGY
The study involved experimental allodynia in female Wistar rats, which were maintained at controlled room temperature and maintained under a 12-hour light/dark cycle. Allodynia was induced by Kim and Chung surgery, with the left L5 and L6 spinal nerves exposed and tightly ligated. Cisplatin-induced neuropathy was induced by repeated intraperitoneal injections of cisplatin (0.1 mg/kg) for 15 days, every third day. Allodynia was evaluated 15 days after the first administration of cisplatin.
Tactile allodynia was determined using the up-down method, with rats exhibiting motor deficiency discarded from tactile allodynia evaluation. The 50% withdrawal threshold of the paw rat was evaluated in a temporal course of 8 hours. A blind design was used, and the area under the curve (AUC) was constructed from the temporal course using the trapezoidal method. The percentage of maximum possible effect (%MPE) was calculated using the following equation:
2.2.2 In silico studies were performed to crystallize structures of the CB1, CB2, and TRPV1 receptors. Crystallized structures were manually curated using Maestro 12.1 with the Protein Preparation Wizard from the Schrodinger suite 2019-424. Docking procedures were performed with Glide 8.4 with the SP methodology and the OPLS3e forcefield.
The isolated organ assay was conducted on a Krebs-Henseleit modified solution, where animals were administered sodium pentobarbital injection and a segment of the middle thoracic esophagus was dissected and placed into the Krebs-Henseleit solution at room temperature. The mechanical activity on the esophagus muscle was recorded using an organ bath filled with Krebs solution and an isometric force transducer. The contractile responses were registered during 10 minutes and taken as control. To test the effect of capsazepine, capsaicin or PhAR-DBH-Me were added to the same nerve segment. Data were processed from the area under the curve (AUC) of the temporal course of contractile response, which was calculated as follows:
%E Drug = (compound gf × 100%E)/ (vehicle or control gf). The contractile effect was measured as grams force (gf) observed as the spike height in the polygraph.
To determine the antiallodynic effect of PhAR-DBH-Me, neuropathic rats were administered either vehicle (saline solution with 10% of Tween 20) or increased logarithmic doses of PhAR-DBH-Me (3.2, 10, 32, and 100 mg/kg). Pregabalin was used as a positive control. In silico studies were performed to characterize the compound’s nature and conduct in vivo assays using various pharmacological tools.